Search Thermo Fisher Scientific
Overview of Detection Probes
Western blotting, enzyme-linked immunosorbent assay (ELISA) and other assay techniques depend on probes that are detectable via chemical tags of one sort or another. Antibodies are the most common basis for designing specific probes. Radioactive isotopes, enzymes and fluorescent dyes are different types of chemical tags that have been used to make probes detectable. This article provides an overview of the types and features of detectable probes.
Introduction
Most analysis methods, including western blotting and enzyme-linked Immunosorbent Assay (ELISA), that are designed to measure the presence or quantity of specific proteins or other molecules in biological samples depend on the use of target-specific probes that are detectable via chemical tags or labels. Antibodies are the most common type of probe; their binding affinity for particular antigens enable those targets to be "found" and detected in a complex sample. However, antibodies are themselves proteins, and they are not specifically detectable in an assay system unless they are tagged for visualization or secondarily probed with another molecule that is tagged.
Different types of chemical labels or tags can be conjugated to secondary or primary antibodies and other molecules to facilitate their visualization (i.e., detection and measurement) by various methods. Radioisotopes were used extensively in the past, but they are expensive, have a short shelf-life, offer no improvement in signal: noise ratio and require special handling and disposal. Enzymes and fluorophores have largely replaced radioactive isotopes as detectable tags for assays. A number of advancements in reagents and instrumentation make these newer technologies more versatile and powerful. Enzymatic tags such as horseradish peroxidase (HRP) are most commonly used for blotting, immunoassays and immunohistochemistry methods. Variants of the bioluminescent enzyme luciferase are also increasingly used for in vivo detection, cell viability assays and reporter gene assays. Fluorescent tags are used predominately for cellular imaging, nucleic acid amplification and sequencing and microarrays; however, fluorescence technology is developing rapidly for application in all types of assays. The representative examples below illustrate how antigen-specific antibodies may be used for chromogenic or fluorescence IHC detection.


Immunohistochemical detection using chromogenic and fluorescently labeled antibodies. For the detection of vascular endothelial zinc finger (VEZF) protein paraffin-embedded brain tissue was processed and probed with a rabbit anti-VEZF polyclonal primary antibody. An anti-rabbit IgG secondary antibody labeled with HRP and the red-precipitating HRP substrate 3-amino-9-ethylcarbazole (AEC) were used for detection. The red colored regions and fibers represent the locations of VEZF (Left). To detect cytokeratin 18 in human colon carcinoma, tissue sections were incubated with a biotinylated anti-cytokeratin 18 antibody and then detected using a Thermo Fisher streptavidin-DyLight 633 conjugate (21844, red fluorescence). Thermo Fisher Hoechst stain (e.g. 33342) was used to counterstain the cell nuclei (blue fluorescence) (right).
An antibody that recognizes the target antigen is called the "primary antibody." If the antibody is labeled with a tag, direct detection of the antigen is possible. Usually, however, the primary antibody is not labeled for direct detection. Instead a "secondary antibody" that has been labeled with a detectable tag is applied in a second step to probe for the primary antibody, which is bound to the target antigen. Thus, the antigen is detected indirectly. The following video provides an example of how antibodies may be used to perform direct and indirect ELISAs.
Watch this video to learn more about ELISA methodologies
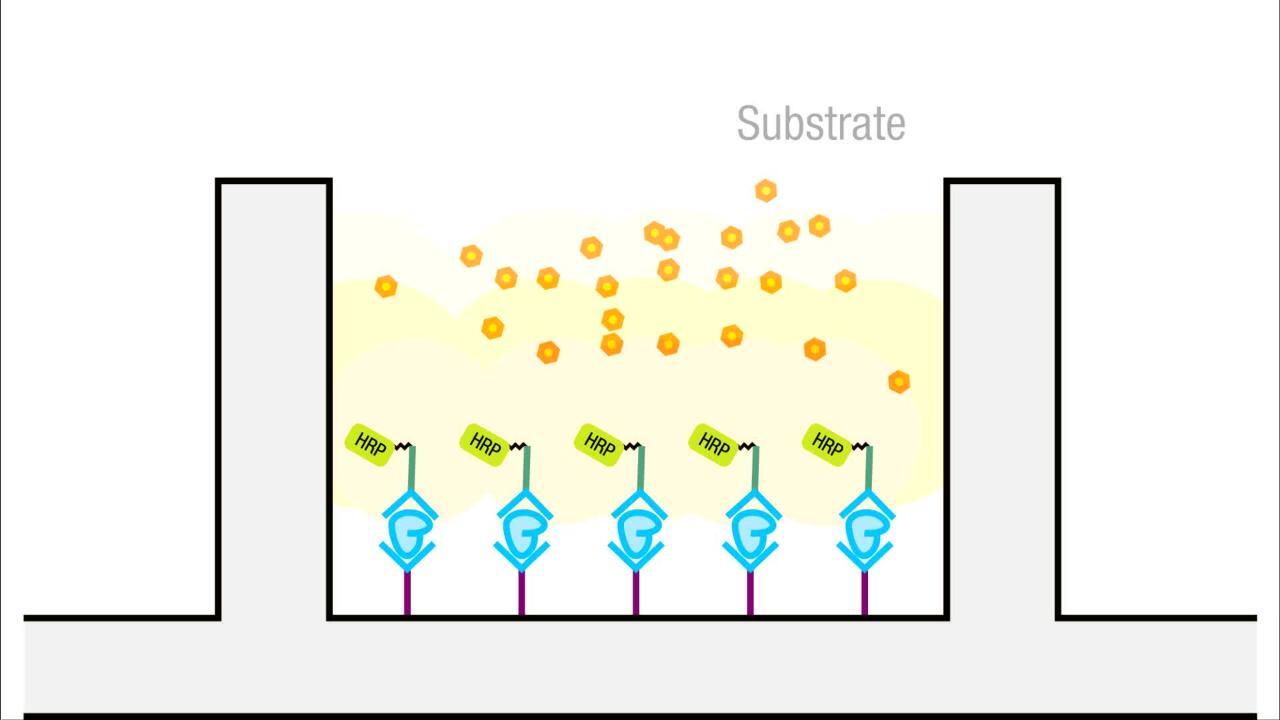
Another form of indirect detection involves using a primary or secondary antibody that is labeled with an affinity tag such as biotin. Then a secondary (or tertiary) probe, such as streptavidin that is labeled with the detectable enzyme or fluorophore tag, can be used to probe for the biotin tag to yield a detectable signal. Several variants of these probing and detection strategies exist. However, each one depends on a specific probe (e.g., a primary antibody) whose presence is linked directly or indirectly to some sort of measurable tag (e.g., an enzyme whose activity can produce a colored product upon reaction with its substrate).

Annotated diagram of immunoglobulin structure. Heavy and light chains are held together by a combination of non-covalent interactions and covalent interchain disulfide bonds, forming a bilaterally symmetric structure. The V regions of H and L chains comprise the antigen-binding sites of the immunoglobulin (Ig) molecules. Each Ig monomer contains two antigen-binding sites and is said to be bivalent. The hinge region is the area of the H chains between the first and second C region domains and is held together by disulfide bonds. This flexible hinge (found in IgG, IgA and IgD, but not IgM or IgE) region allows the distance between the two antigen-binding sites to vary. Also shown are several functional groups that are selectable targets for practical bio conjugation.
Protein assay technical handbook
This 60-paged technical handbook and product guide will help you to select an appropriate assay method based on assay time, sensitivity, compatibility, standard curve linearity, and protein-to-protein variation. Learn about our wide range of colorimetric (copper or dye-based) and fluorescent protein assays, as well as our more specialized assays to quantify peptides, antibodies, protein modifications, or functional (enzymatic) classes of proteins. Discover tools and strategies to help optimize your protein quantitation assays to ensure more accurate downstream results.

Learn more
- Primary Antibodies
- Introduction to Secondary Antibodies
- Immunoglobulin Structure and Classes
- Overview of Western Blotting
- Overview of ELISA
- Overview of Immunohistochemistry
- Avidin-Biotin Interaction
- Antibodies Learning Center
- Antibodies Support Center
- Protein Assays and Analysis Support Center
- Guide to Primary Antibodies
- Invitrogen Antibody Validation
- Antibodies and Protein Information
Primary antibodies as probes
Thousands of primary antibodies are commercially available for protein targets with a history of investigation in biological research. Except for a few very popular research targets, these primary antibodies are offered without detectable tags, and some sort of secondary (indirect) detection method is required. Nevertheless, nearly any antibody can be labeled with biotin, HRP enzyme or one of several fluorophores if needed.
Depending on the application to be performed, different levels of purity and types of specificity are needed in a supplied primary antibody. To name just a few parameters, antibodies may be monoclonal or polyclonal, supplied as antiserum or affinity-purified solution, and validated for native protein or denatured protein detection.
If no antibodies exist for an antigen of interest, new antibodies can be produced (raised) using well established techniques for immunizing animals with prepared forms of the antigen. A variety of reagents are available to assist in antibody production and purification, and various companies specialize in antibody production services.
On demand webinar:
Addressing the antibody reproducibility crisis: A panel discussion with key scientific leaders
- Aled Edwards, PhD, CEO, Structural Genomics Consortium, University of Toronto, Toronto, Ontario, Canada; International Working Group for Antibody Validation (IWGAV) member
- Matt Baker, Director of Strategy and Partnering for Antibodies and Immunoassay, Biosciences Division, Thermo Fisher Scientific
- Anita Bandrowski, PhD, Scientific Lead, Neuroscience Information Framework, Center for Research in Biological Systems, University of California at San Diego, San Diego, California, USA; Founder and CEO of SciCrunch
- Paul K. Wallace, PhD, Professor of Oncology, and Director, Flow and Image Cytometry Facility, Roswell Park Cancer Institute, Buffalo, NY, USA; Associate Professor of Pathology, State University of New York at Buffalo, NY, USA
- John Rogers, PhD, Senior R&D Manager, Mass Spectrometry Reagents, Protein and Cell Analysis, Biosciences Division, Thermo Fisher Scientific
- Christoph Hergersberg, PhD, Vice President of R&D for Protein and Cell Analysis, and Antibodies and Immunoassays, Biosciences Division, Thermo Fisher Scientific
Abstract
Every year, millions of dollars are wasted on poorly characterized and performing antibodies. Key researchers in the antibody community have recently come together to address this antibody crisis and develop standards to ensure proper characterization and consistency for antibodies in the laboratory. Join us for a panel discussion with members of the research community and Thermo Fisher Scientific leaders, as they discuss the antibody reproducibility crisis and proposed testing standards for antibodies to ensure they’re binding to the intended targets.

Secondary antibodies as probes
Most primary antibodies are produced in mouse, rabbit or one of several other species. Nearly all of these are antibodies of the IgG class. Therefore, it is relatively easy and economical for manufacturers to produce and supply ready-to-use, labeled secondary antibodies for most applications and detection systems.
Even so, several hundred options are available, differing in the level of purity, IgG- and species-specificity, and detection label. The choice of secondary antibody depends upon the species of animal in which the primary antibody was raised (the host species). For example, if the primary antibody is a mouse monoclonal antibody then the secondary antibody must be an anti-mouse antibody obtained from a host other than the mouse.

Direct and indirect protein detection. The surface on which the target protein is bound and immobilized is either a membrane (Western blot) or a microplate well (ELISA).
Learn more
Select products
Biotin-binding proteins as probes
The highly specific affinity interaction between biotin (a small vitamin molecule) and Avidin, Streptavidin, or NeutrAvidin proteins is the basis for many kinds of detection and affinity-purification methods. Biotin is very small (244 Daltons), so its covalent attachment to antibodies or other probes rarely interferes with their functions. Yet its presence as a tag on a probe allows efficient and specific secondary detection with either Avidin, Streptavidin, or NeutrAvidin. These biotin-binding proteins are available in purified forms labeled with enzymatic or fluorescent tags that enable detection in many kinds of assays systems.
Many biotinylation reagents and kits are available that allow efficient and stable labeling of nearly any kind of antibody, protein or other macromolecule. Even probing systems (such as EMSA, Enzyme Mobility Shift Assay) that are not based on antibodies can be adapted for detection using Avidin-biotin chemistry. In addition, biotin systems have several features that enable signal amplification to yield high sensitivity.
Avidin, Streptavidin, and NeutrAvidin bind very strongly and specifically to biotin. However, each protein has their limitations in certain assays. Avidin, is glycosylated which may lead to nonspecific lectin binding in assays. However, Streptavidin contains a RYD motif, a bacterial recognition sequence, which can cause background binding with certain samples. Another alternative is to use NeutrAvidin protein which is an exclusive, deglycosylated form of Avidin that avoids the drawbacks of both native Avidin and Streptavidin.

Schematic of the Avidin-biotin interaction. Avidin (or Streptavidin or NeutrAvidin) can bind up to four biotin molecules, which are normally conjugated to an enzyme, antibody or target protein to form an Avidin-biotin complex. † denotes that Avidin is also often conjugated to an antibody, target protein or immobilized support
Learn more
- Avidin-biotin Systems
- Biotinylation
- Tech Tip #16: Block endogenous biotin
Enzyme labels for detection
Enzymatic labels are most commonly used as secondary antibody (or Streptavidin) tags for detection in blotting and immunoassays. Enzymes provide detectable signal via their activity; reaction with a specific substrate chemical yields a colored, light-emitting, or fluorescent product. While reporter enzymes like beta-galactosidase and luciferase have been successfully used to make probes, alkaline phosphatase (AP) and horseradish peroxidase (HRP) are the two enzymes used most extensively as labels for protein detection. An array of chromogenic, fluorogenic and chemiluminescent substrates is available for use with either enzyme.
Alkaline phosphatase, usually isolated from calf intestine, is a large (140 kDa) protein that catalyzes the hydrolysis of phosphate groups from a substrate molecule resulting in a colored or fluorescent product or the release of light as a byproduct of the reaction. AP has optimal enzymatic activity at a basic pH (pH 8-10) and can be inhibited by cyanides, arsenate, inorganic phosphate and divalent cation chelators, such as EDTA. As a label for western blotting, AP offers a distinct advantage over other enzymes. Because its reaction rate remains linear, detection sensitivity can be improved by simply allowing a reaction to proceed for a longer time period.
Horseradish peroxidase is a 40 kDa protein that catalyzes the oxidation of substrates by hydrogen peroxide, resulting in a colored or fluorescent product or the release of light as a byproduct of the reaction. HRP functions optimally at a near-neutral pH and can be inhibited by cyanides, sulfides and azides. Antibody-HRP conjugates are superior to antibody-AP conjugates with respect to the specific activities of both the enzyme and antibody. In addition, its high turnover rate, good stability, low cost and wide availability of substrates makes HRP the enzyme of choice for most applications. Because of the small size of the HRP enzyme, further increases in sensitivity may be achieved by using poly-HRP conjugated secondary antibodies and may eliminate the need for using ABC type amplification systems for some researchers.

Common ELISA formats. In the assay, the antigen of interest is immobilized by direct adsorption to the assay plate or by first attaching a capture antibody to the plate surface. Detection of the antigen can then be performed using an enzyme-conjugated primary antibody (direct detection) or a matched set of unlabeled primary and conjugated secondary antibodies (indirect detection).
Learn more
Select products
Luciferase reporters for bioluminescence detection
Although luciferase enzymes could be grouped into the enzymatic labels section, the scope that these proteins are used for biomedical research is more diverse than that of other enzymatic labels. Bioluminescent light emitted from luciferase differs from fluorescent light, in that the light is a product of an enzymatic reaction rather than the release of energy that is initially absorbed by a fluorescent molecule. Because luciferase enzymes are bioactive proteins, they can be expressed in cells or even live animals to detect proteins, small molecules or gene expression in real time.
Luciferase enzymes have been isolated from a large number of animal species that use them for defense, camouflage, mating and feeding, and each species-specific luciferase has distinct characteristics that provide flexibility for use in biological assays. These characteristics include size, cofactor requirements (Mg, ATP), substrate (D-luciferin, coelenterazine), light emission spectra reaction kinetics and whether or not the enzyme is secreted. These characteristics provide a wide range of detection sensitivities and emission time to accommodate different single- and multiplex experimental designs.

Firefly luciferase bioluminescent reaction. This light-emitting enzymatic reaction is required for the bioluminescence of fireflies and click beetles. Firefly luciferase catalyzes the oxidation of firefly luciferin. The reaction is both oxygen and ATP dependent.
Learn more
Select products
Fluorescent labels for detection
Historically, fluorophore-labeled secondary antibodies and other probes were used in a small number of cell biology applications such as flow cytometry (FC), fluorescence-activated cell sorting (FACS) and immunohistochemistry (IHC) using fluorescence microscopy. Until recently, the two most common fluorophores for labeling probes were fluorescein (fluorescein isothiocyanate, FITC) and rhodamine (tetramethyl rhodamine isothiocyanate, TRITC). Other labels include fluorescent proteins such as the various forms of green fluorescent protein (GFP) and the phycobiliproteins (allophycocyanin, phycocyanin, phycoerythrin and phycoerythrocyanin). While having the ability to produce an intense fluorescent signal for detection, fluorescent proteins can be difficult to optimize for conjugation purposes and may create steric hindrance or background signal issues in binding assays.
The use of fluorophore-conjugated probes in blotting and immunoassays requires fewer steps compared to the use of enzymatic labels because there is no substrate development step to perform. While the protocol is shorter, fluorescent detection requires special equipment and the sensitivity is not a high as that which can be obtained with enzymatic chemiluminescent systems. Although not as sensitive as enzymatic detection, fluorescent detection methods reduce chemical waste and have the added advantage of multiplex compatibility (using more than one fluorophore in the same experiment).
The growing demand for multiplex assays has driven the development of many new fluorescent dyes. These new fluorophores are brighter and more photo stable than the traditional fluorescein and rhodamine molecules and comprise a broader range of non-overlapping spectra. Together with the advances in the digital imaging equipment, particularly infrared and near-infrared imaging, these new fluorophores enable extremely powerful analyses in all types of protein detection techniques. In the following example, immunofluorescence methods were used to detect the Glucocorticoid receptor in a cancer cell line.

Immunofluorescence detection of the Glucocorticoid receptor. The Glucocorticoid receptor was detected in HeLa cells using the Glucocorticoid Receptor Monoclonal Antibody (BuGR2). Glucocorticoid receptor (green), F-Actin staining with Phalloidin (red) and nuclei with DAPI (blue) are shown. Cells were probed without (control) or with an antibody recognizing Glucocorticoid receptor according to the product sheet protocol, washed with PBS and incubated with a DyLight-488 conjugated secondary antibody (Product # 35552 for GAR, Product # 35503 for GAM).
Recommended reading:
- Harlow, E. and Lane, D. (1988). Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
- Malik, V.S. and Lillehoj, E.P. (1994). Antibody Techniques. Academic Press, Inc., San Diego, CA.
- Green N.M. (1963). Avidin. 3. The Nature of the Biotin-Binding Site. Biochem J.89:599–609.
- Livnah, O; Bayer, EA; Wilchek, M; Sussman, JL (1993). "Three-dimensional structures of Avidin and the Avidin-biotin complex". Proceedings of the National Academy of Sciences of the United States of America. 90(11): 5076–80.
- Green N.M. (1963). Avidin. 3. The Nature of the Biotin-Binding Site. Biochem J.89:599–609.
- Sambrook J. and Russell D. W. (2001) Molecular cloning: A laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press.
- Keilin D. (1966) The history of cell respiration and cytochrome. Cambridge, Cambridge U.P. xx, 416.
- Negrin R. S. and Contag C. H. (2006) In vivo imaging using bioluminescence: A tool for probing graft-versus-host disease. Nat Rev Immunol. 6, 484-90.
- Smith K. C. (1989) the science of photobiology. New York: Plenum Press. Viii, 426.
- Campbell A. K. and Herring P. J. (1990) Imidazolopyrazine bioluminescence in copepods and other marine organisms. Marine Biology. 104, 219-25.
- Robison B. H. and Young R. E. (1981) Bioluminescence in pelagic octopods. Pacific Science. 35, 39-44.
- Cazes J. (2001) Encyclopedia of chromatography. New York: Marcel Dekker. Xxx, 927 p.
- Chalfie M. et al. (1994) Green fluorescent protein as a marker for gene expression. Science. 263, 802-5.
仅供科研使用,不可用于诊断目的。